This web page was produced as an assignment for Genetics 564, an undergraduate capstone course at UW-Madison.
What is Phylogeny?
Phylogeny is the study of the evolutionary relationships among organisms by considering inherited characteristics of species and other information known about species[1]. These relationships can be represented with phylogenetic trees which illustrate the relatedness and evolutionary development between species - much like a family tree!
What are Phylogenetic Trees?
Phylogenetic trees represent the evolutionary relationships among a group of organisms based on shared derived traits coming from a most common ancestor. The comparison and analysis of a combination of external morphology, internal anatomy, behaviors, biochemical pathways, DNA and protein sequences, and fossils can be used to build phylogenetic trees [2].
Phylogenetic trees are hypotheses. The analysis of different combinations of shared characteristics between organisms can show multiple ways in which organisms relate to one another. And as research keeps advancing, phylogenetic trees are frequently updated and revised to disclose the current knowledge. The most common methods for building phylogenetic trees are neighbor joining, maximum likelihood, and average distance.
Phylogenetic trees are hypotheses. The analysis of different combinations of shared characteristics between organisms can show multiple ways in which organisms relate to one another. And as research keeps advancing, phylogenetic trees are frequently updated and revised to disclose the current knowledge. The most common methods for building phylogenetic trees are neighbor joining, maximum likelihood, and average distance.
Building Phylogenetic Trees
1. Compile sequence data for homologs.
2. Align sequences using software like ClustalWOmega or MEGA.
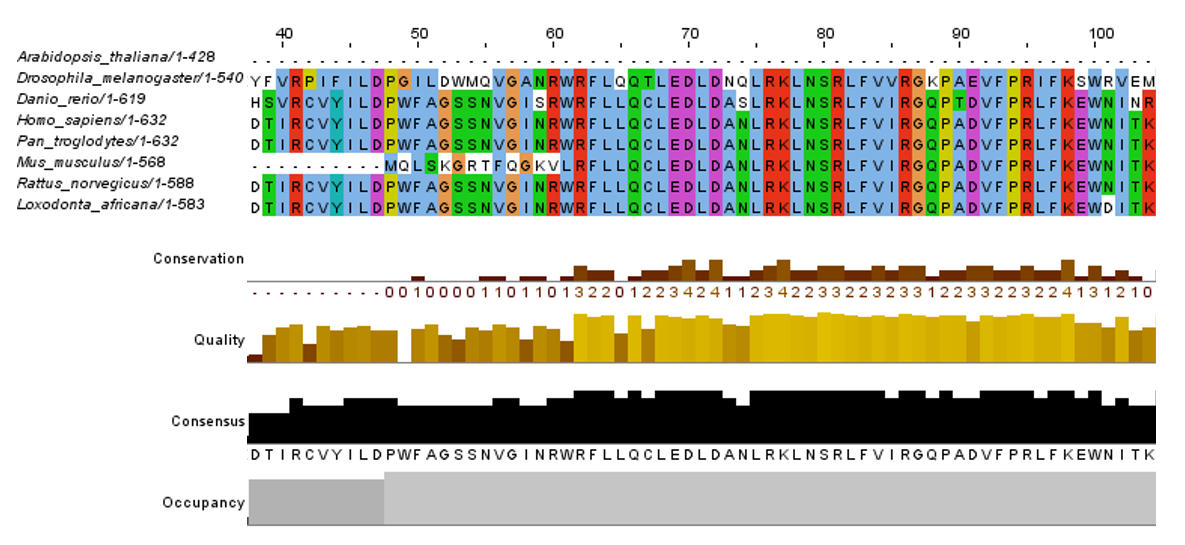
Cry1 protein sequence alignment with ClustalW algorithm.
3. Create phylogenetic trees using MEGA, choosing method that best fits interest. [3]
Maximum Likelihood
For this method, different trees are formed by grouping different organisms with data from observed character traits. The initial trees are then compared and given likelihood scores. Higher likelihood scores relate to a higher probability that characters observed developed in that way over time. Data considered for maximum likelihood includes sequence alignments.
Neighbor-Joining
Neighbor-joining is a fast distance based method that requires the development of a matrix specifying the magnitude of relatedness and amount of differences between sequences of each pair of taxa or species. Branches are formed as this method joins sequences that are most similar into nodes and repeats this process until all compared species are included in the tree.
Average Distance
This method uses relatedness scores to join species most closely related with equal branch lengths. Methods like neighbor-joining and maximum likelihood can be used to build the tree and using average distance branch lengths can be adjusted.
Maximum Likelihood
For this method, different trees are formed by grouping different organisms with data from observed character traits. The initial trees are then compared and given likelihood scores. Higher likelihood scores relate to a higher probability that characters observed developed in that way over time. Data considered for maximum likelihood includes sequence alignments.
Neighbor-Joining
Neighbor-joining is a fast distance based method that requires the development of a matrix specifying the magnitude of relatedness and amount of differences between sequences of each pair of taxa or species. Branches are formed as this method joins sequences that are most similar into nodes and repeats this process until all compared species are included in the tree.
Average Distance
This method uses relatedness scores to join species most closely related with equal branch lengths. Methods like neighbor-joining and maximum likelihood can be used to build the tree and using average distance branch lengths can be adjusted.
CRY1 Protein Phylogeny
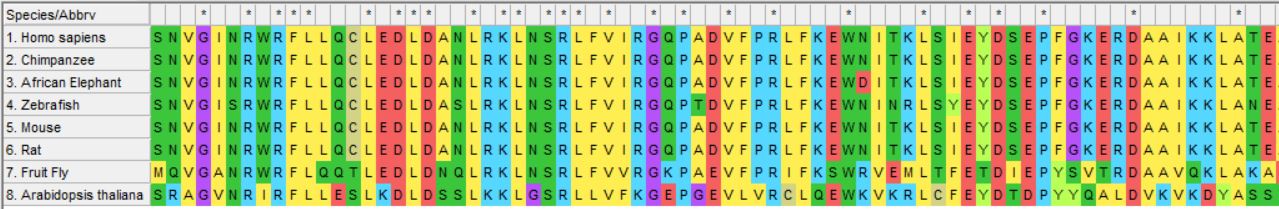
Figure 1. Cry1 protein sequence alignment with MEGA algorithm.
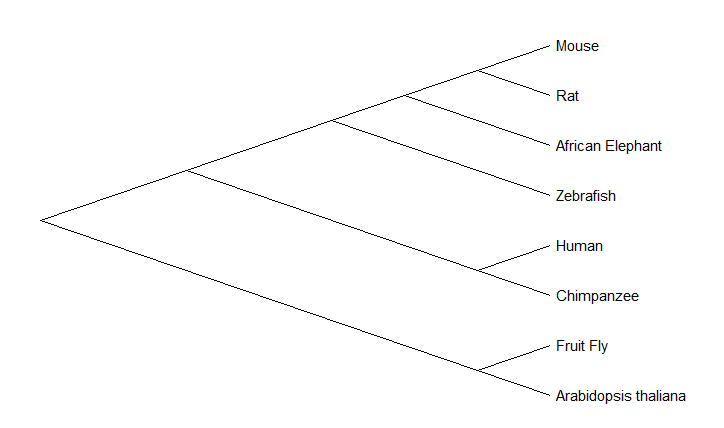
Figure 2. Cry1 protein Maximum Likelihood phylogenetic tree built with MEGA.
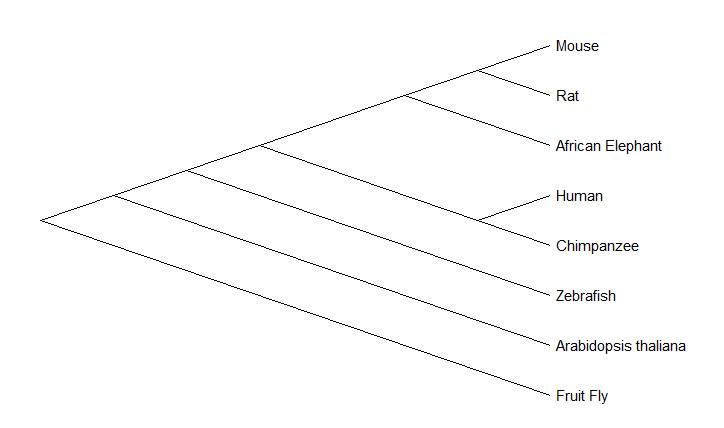
Figure 3. Cry1 protein Neighbor-Joining phylogenetic tree built with MEGA.
Conclusion
The maximum likelihood phylogenetic tree deviated from the findings of the neighbor-joining phylogenetic tree in the localization of the diverging event of the CRY1 protein between zebrafish and mammals. Based on prior knowledge concerning the diversification of vertebrates and appearance of mammals, the neighbor-joining phylogenetic tree showcases more efficient relationships between the organisms' protein.
Proximity of zebrafish and mice to humans, make them both good candidates for studying CRY1.
Proximity of zebrafish and mice to humans, make them both good candidates for studying CRY1.
References.
1. Berkeley University. (n.d.) The family tree. Retrieved from https://evolution.berkeley.edu/evolibrary/article/0_0_0/evo_04[2] EMBL-EBI. (n.d.)
2. Phylogenetic trees. (n.d.). Retrieved from https://www.khanacademy.org/science/biology/her/tree-of-life/a/phylogenetic-trees
3. Barton, N. H. (2007). Evolution. Retrieved from http://evolution-textbook.org/content/free/contents/ch27.html#ch27-4-2
Header image.
1. Berkeley University. (n.d.) The family tree. Retrieved from https://evolution.berkeley.edu/evolibrary/article/0_0_0/evo_04[2] EMBL-EBI. (n.d.)
2. Phylogenetic trees. (n.d.). Retrieved from https://www.khanacademy.org/science/biology/her/tree-of-life/a/phylogenetic-trees
3. Barton, N. H. (2007). Evolution. Retrieved from http://evolution-textbook.org/content/free/contents/ch27.html#ch27-4-2
Header image.
|
Contact me
Sara Acosta Villarreal Genetics and Genomics, UW-Madison [email protected] Last updated: May 10, 2019 |
Visitors Worldwide
|
